|
UniformMu Transposon Resource
Downloads:
W22 to B73 cross-reference:
Excel spreadsheet Genomic coordinates for Zm-B73-REFERENCE-GRAMENE-4.0 (aka B73 RefGen_v4): Release 9 Excel spreadsheet Release 9 Excel spreadsheet with gene structure List of gene models from the B73 RefGen_v3 Filtered Gene Set that have UniformMu insertions: Release 8 Excel spreadsheet List of gene models from the B73 RefGen_v2 Filtered Gene Set that have UniformMu insertions including 100 bp upstream or downstream: Release 7 Excel spreadsheet Release 8 Excel spreadsheet List of gene models from the B73 RefGen_v2 Filtered Gene Set that have UniformMu insertions in exons: Release 7 Excel spreadsheet Release 8 Excel spreadsheet All UniformMu downloads, including flanking sequence for B73 and W22.
Genome alignments The UniformMu Resource in Detail
I. Key features and current status of the UniformMu resource
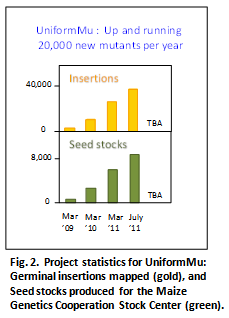
(March, 2011) UniformMu is a special maize population developed specifically for genetics research. A key feature is that genetic variation between individual UniformMu plants is limited to mutations caused by Robertsons Mutator transposons (McCarty et al., 2005). This is achieved by introgression of active Mu transposons into agenetically uniform W22 inbred strain. UniformMu maize is widely used for experimental analysis of gene function because 1) the uniformity of the plants and seeds provides an excellent basis for phenotype comparisons (uniform wild type controls), and 2) mutations of individual genes are caused by Mu transposable element insertions. Gene function can then be studied at the biochemical, metabolic, and whole-plant levels as a consequence of the specific changes in gene sequence. Mutations in genes are identified by a sequence-based mapping of germinal Mu insertions in the maize genome and seeds are available to researchers (via MaizeGDB and Maize Coop Stock Center).
Key features:
The latest UniformMu data release at MaizeGDB (March 2011) includes 26,211 germinal insertions mapped in 5,127 seed stocks, each available immediately and free of charge from the Maize Genetics Cooperation Stock Center. By July, 2011, we will have mapped up to 38,000 germinal, heritable insertions in 8,256 seed stocks. The collection currently contains insertions in an estimated 31% of maize genes based on mapped Mu sites in at least 12,127 high-confidence maize genes in the Filtered Gene Set (v 5b.60, MaizeSequence.org). These genes also include 5,329 that have multiple insertion alleles invaluable for confirming genotype-phenotype relationships (Table 1).
II. Finding insertions at MaizeGDB.org
III. Recommended care and handling of UniformMu insertion lines The envelope will contain pooled seed from sib-pollinated F3 plants (see UniformMu Methods). The F3 lines are sibbed to minimize selection against insertions that may have deleterious phenotypes (e.g. lethal’s, male steriles, etc). Many interesting, but deleterious mutations are thus preserved as heterozygous insertions; whereas, roughly half of the Mu insertions in UFMu lines are homozygous and will be present in all plants. Even if the Mu-insert of interest is deleterious, it should still be carried by at least some heterozygous F3 seed in the sample provided. -- We recommend planting at least eight seeds in the field or greenhouse such that all plants can be sampled for DNA and pollinated.
-- We also recommend concurrently planting W22 wild type seeds
(available from the Stock Center &151; catalog # X17EA), so that these can
be used as additional mothers for pollen from mutant plants (Additional
W22 can also be planted about 10 days before and after the mutant seed
to increase potential for silk availability at the time of mutant
anthesis). This is important for three reasons. First, although
UniformMu lines generally have good fertility, some may carry mutations
that reduce vigor and fecundity (e.g. male sterility). Having wild type
pollen and silks available helps ensure recovery of seed from the
selected UFMu line. Second, backcrosses to wild type are often a
necessary step towards developing a family of segregating progeny,
where wildtype and mutant siblings can be tested for co-segregation of
any phenotypic features with presence of the Mu-insert of interest.
This is especially important if the insert is homozygous in the UFMu
line, in which case this generation will be needed to generate a
suitable segregating family. Third, the UFMu lines typically carry 5
to 10 novel insertions (sometimes more), so a backcross will aid
separation and independent assortment of these in subsequent
generations (A given phenotype in the first generation may or may not
be due to the Mu-insert of interest). Recommended backcrosses can thus
speed analyses. PCR confirmation of Mu insertions.
IV. UniformMu Methods - genetics, sequencing and bioinformatics
Field genetics - Production of F3, UniformMu seed stocks for sequence-indexing and public distribution. To isolate new mutations, plants heterozygous for a highly active MuDR (i.e. self-pollinated ears that segregate 3:1 for densely spotted, bz1-mum9 andstable bronze kernels) are backcrossed to our standard W22 ACR inbred strain (McCarty et al., 2005). The resulting F1 plants are grown, self-pollinated and screened for stable bz1 seed (Fig 4; McCarty et al., 2005). Because the F1 plants are heterozygous for MuDR, the self-pollinated ears segregate Mu-inactive seed that havea stable bronze (non-spotted) kernel phenotype. The stable bronze F2 seed are grown and self-pollinated to establish confirmed homozygous MuDR(-), Mu-inactive derivative lines. Twenty, spot-free F3 kernels are selected from each line, grown and the plants sib-pollinated. To create a seed stock suitable for sequence analysis and public distribution through the Stock Center, a balanced pool of 200-300 seed is prepared from 2-5 ears (depending on ear size, and quality) from each family. To ensure highest possible seed quality all seed stocks are screened manually by experienced personnel (including students, mentors, and technical staff to eliminate residual Mu-activity, cracking, mold and other defects that may impact seed viability. Maize plants are grown and pollinated at the UF PSREU experimental farm at Citra, FL and at the Gray Research facility located in Askum, Ill. The Florida location allows two generations per year while the relatively mild central Illinois summer season is optimal for production of high quality seed stocks. Sequence-indexing of Mu insertions by Illumina sequencing. We have developed a highly efficient protocol for sequencing Mu flanking sequences using the Illumina platform (Illumina.com). Plants are grown in 24 X 24 grids (576 lines per grid) creating a 2D array of 48 pooled DNA samples (24 for each axis). The 48 DNA samples are sequenced in a single multiplexed library using one lane of an Illumina flow cell. Multiplexing is implemented using a custom set of Illumina Flow Cell A sequencing adaptor oligos each incorporating one of 64 different 4-base key codes into the 5'-end of the sequence. The fourth base of the code is a check sum that allows detection of sequencing errors in the multiplex key. To eliminate chimera PCR artifacts that can result in "key swapping" among the multiplex samples, all steps of library construction are performed eparately for each of the 48 DNA samples and sub-libraries are pooled prior to the cluster generation stage in the Illumina sequencing protocol. In brief, to obtain unbiased sampling of the genome, each DNA sample is randomly sheared using a BioRuptor sonication instrument, end-polished and ligated to a universal adaptor oligonucleotide (Fig 5). PCR I is performed using a Mu TIR specific primer and the universal adaptor primer. A nested PCR II reaction is then performed to incorporate the Illumina A and B flow-cell adaptor primers into Mu specific amplicons. The 3' half of the long Illumina A-sequencing adaptor is fused to a nested Mu TIR specific oligo that is anchored six bases from the 3'-end of the TIR (Fig 5a), and the B-adaptor sequence is incorporated by fusion to the universal adaptor primer. The six bases of TIR in the genomic sequence downstream of the A-primer are used to confirm authentic Mu flanking PCR products. Less than 1% of reads are rejected for mispriming. The PCR II products are size-selected by excision from agarose gels where each sample is run in a separate gel to eliminate the possibility of cross-contamination. Incorporation of the 5-prime portions of the A-sequencing adaptor is completed in a third PCR reaction. All PCR steps use a minimum number of cycles to mitigate PCR bias.
Bioinformatics analysis and mapping of Mu flanking sequences.
In a typical 72 base run performed at NCGR (Santa Fe, NM), our custom
multiplex libraries yield 25-32 million reads per lane providing
greater than 500,000 quality trimmed reads per multiplex DNA sample.
With current Illumina technology, 72-base runs return high yields of
reads conducive to statistically robust axis assignments while allowing
relatively fast turn-around due to availability of 72-base lanes at
sequencing centers (e.g. NCGR). Longer reads may be obtained from the
same libraries without modification. After parsing of the key code,
trimming of the Mu TIR sequence, reads retain 38 bases of precisely
anchored genomic DNA sequence flanking the Mu insertion (Fig 5 b). The
trimmed reads are mapped to the B73 maize genome (AGP v2
pseudomolecules) using BLASTN run in parallel on the UF HPC computing
cluster (www.hpc.ufl.edu). Using 64 processors, mapping of 32 M reads is
completed in about 2 hours. The BLAST output is parsed into a database
of mapped Mu insertion sites using a cutoff BLAST expectation score of
10-7. Flanking sequences that map within 3 bp in the maize genome are
assigned to the same insertion locus. For each insertion locus, read counts in each grid axis are used to locate maize lines in grid that contain that insertion (Fig 6). Insertion loci that map to low copy locations in the genome (maximum of 4 unresolved copies; >95% are single copy) and that have unambiguous, statistically robust assignments (axis counts with a C2 score less than 10-5) to individual lines in the grid are assigned a unique six digit identifier (e.g. mu103480) and submitted to MaizeGDB.org. In parallel, seed stocks (minimum of 200 high-quality seed per line) for each grid are deposited in the Stock Center. Consensus flanking sequences for each insertion are assembled from the Illumina reads using the AMOS simple consensus program (Meyers et al., 2000) and paired flanking sequences are deposited in Genbank as gapped 76 bp sequences (or longer depending on Illumina read length).
V. Search POPcorn for UniformMu seed stock References McCarty DR, Meeley RB. (2009) Transposon resources for forward and reverse genetics in maize. In. Handbook of Maize: genetics and genomics. (Ed. JL Bennetzen, S. Hake). Springer Science Berlin. pp 561-584. McCarty DR, Settles AM, Suzuki M, Tan BC, Latshaw S, Porch T, Robin K, Baier J, Avigne W, Lai J, Messing J, Koch KE, Hannah LC. (2005) Steady-state transposon mutagenesis in inbred maize. Plant J. 44:52-61. Myers E, Sutton G, et. al. (2000) A Whole-Genome Assembly of Drosophila. Science 287:2196-204 Schnable PS et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112-5 Settles AM, Holding DR, Tan BC, Latshaw SP, Liu J, Suzuki M, Li L, O'Brien BA, Fajardo DS, Wroclawska E, Tseung CW, Lai J, Hunter CT 3rd, Avigne WT, Baier J, Messing J, Hannah LC, Koch KE, Becraft PW, Larkins BA, McCarty DR. (2007) Sequence-indexed mutations in maize using the UniformMu transposon-tagging population. BMC Genomics. 8:116 |


|